
 Avirup Sanyal 1,2, Abhranil Gangopadhayya3 and Som Banerjee4
Avirup Sanyal 1,2, Abhranil Gangopadhayya3 and Som Banerjee4
1- Trivedi School of Biosciences, Ashoka University, Haryana, India
2- National Center for Biological Sciences, Bengaluru, India
3- RA-21, Block-RA, Sector-IV, Salt Lake, Kolkata, India
4- Institute for Virology and Immunobiology, Julius-Maximilians-Universität, Würzburg, Germany
E-mail: avirup.sanyal@ashoka.edu.in
Nature has created a perfect sandbox for its creations to thrive in, which includes the dreaded pathogens causing often-deadly infectious diseases. While we humans are constantly barraged by an array of disease-causing pathogens, sometimes their main host is not us, but other animals, often referred to as ‘reservoirs’. Reservoirs like bats and rodents have been known to harbor a wide array of ‘unknown’ infectious agents. The importance of studying these poorly known agents cannot be underscored enough for tropical countries, wherein exotic diseases are commonplace and ever-emerging.
The focus on studying these wild reservoirs seems to have been re-ignited after the outbreak of the recent SARS-CoV-2 and its possible links to a bat reservoir or an intermediate pangolin host. Fundamental questions such as ‘where did these zoonotic pathogens originate from?’, ‘what makes bats so suitable for coexistence with viruses?’, ‘what triggers the spillover of a certain pathogen out of the blue?’ remain unexplored since research is more geared towards solving the current crisis at hand. However, the inability to find answers to these questions often results in incessant culling of wildlife. If one asks whether disease dynamics can be studied and possible intervention measures undertaken without embracing ecological parameters, the answer is a resounding no.
About the Influenza virus
Influenza viruses are a key member of the family Orthomyxoviridae, characterized by their segmented RNA genomes. Influenza viruses consist of mainly four species – A, B, C & D. All except species D are known to infect humans, among which species A has the most subtypes and thus the highest potential to cause a pandemic due to its ever-evolving nature. The viruses are composed of spherical particles that enclose eight different segments of genetic material in case of species A, that each produce a unique flu virus protein. Since each segment is free of the other and can independently give rise to a protein, it is theoretically possible for any combination of the segments to exist within a particle (Fig.1).
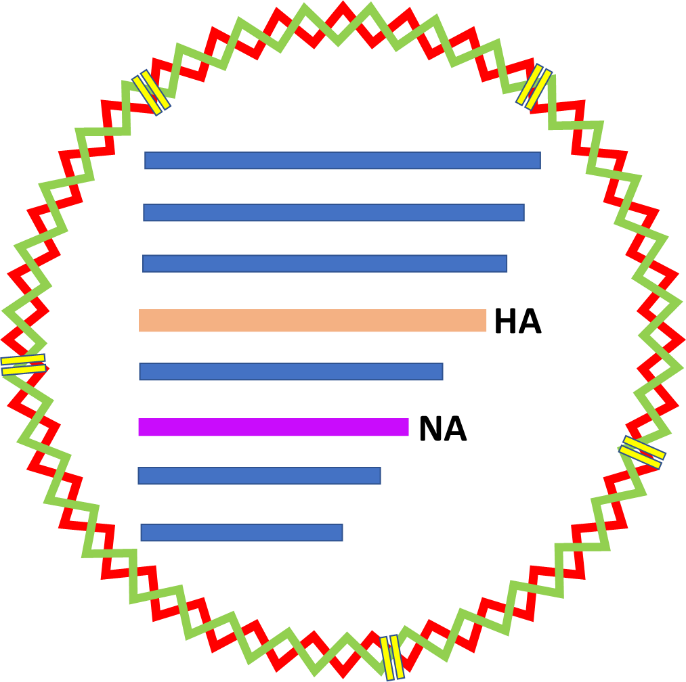
Revisiting the basic concepts of mathematics, permutation and combination, something that we all have come across in our school days (whether we liked it or not), will help us understand this concept of reassortment better. Influenza is characterized by its two prized possessions – the Haemagglutinin (HA) and Neuraminidase (NA) proteins present on its outer surface.
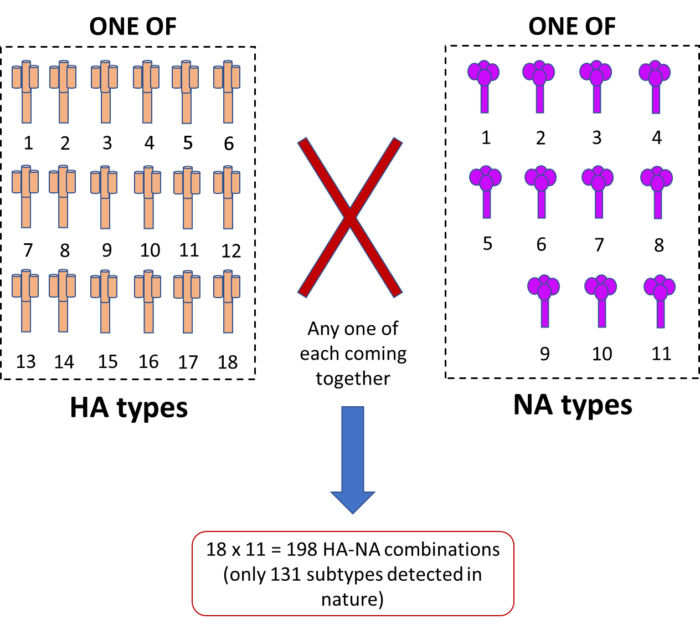
There are 18 different kinds of HA molecules, and 11 different kinds of NA molecules. Now, if we were to arrange them in various combinations of two, the total would amount to 198 possible unique influenza ‘subtypes’. Few of them are known to cause human disease, however, to our great relief! But the virus is made up of eight segments, only two of which are HA and NA, thus allowing any combination of the eight segments to pop up in case multiple influenza subtypes infect the same host body. Thus, the hosts of influenza, swine in particular, are a melting pot for the flu virus to infect, reassort and leave with a fresh new combination of gene segments (Fig.2).
Discovery of two novel bat influenza viruses, H17N10 and H18N11 by Tong et. al in 2012 and 2013 respectively sparked interest in understanding the diversity and reassortment of these unique flu viruses in wild reservoirs. These viruses are distinct from other well-characterized Influenza viruses, and have subsequently captured the attention of the scientific community.
If we look back in history, the H1N1 strain of 1918, which caused the Spanish Flu, reassorted to the H2N2 Asian strain in 1957, followed by the H3N2 strain of Hongkong in 1968, the H1N2 strain of 2001, and finally the triple reassortant pandemic strain H1N1 of 2009.
How the flu virus and animals interact?
Influenza viruses have been documented to infect and reside in a wide range of animals like birds, swine and more. Sialic acid residues, which are modified carbohydrates widely distributed in animal tissues, are the predominant host cell receptors used by the flu virus to infect avian or human cells, but the bat influenza virus makes use of MHC Class II molecules as their host cell receptors in bats. This uncovers potential risk for humans and other species in whom these molecules are indiscriminately present and play a key role in modulating our immune response (Karakus et.al, 2019).
Almost every kind of cell in our body has the MHC molecule on its surface, which can make a person either more or less susceptible to a disease, be it viral or non-viral. This also applies to non-human animals, and MHC types are usually conserved among types of populations. MHCs are grouped into two classes – class I and class II, both having unique functions. In case of bats, it has been hypothesized that the MHC-II molecules allow infection but not to an extent that they fall sick and die, also allowing the virus to persist in the bat and spread further. Since MHC-II molecules are a keystone in adaptive immunity, the response generated in bats may be sufficient to overcome the pathological effects of viral infections, such as influenza, but not enough to clear the virus out from their bodies. It may also be that a virus uses a certain MHC or a similar molecule of immunological importance to aggravate its own stronghold in the body, and thus the resulting symptoms caused by tissue and organ damage.
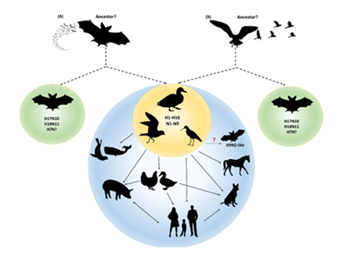
On the other hand, the N11 protein which was initially thought to be dispensable during viral replication and shedding of H18N11 in the bat host, was experimentally shown to be essentially required for transmission between individual Neotropical Jamaican fruit bats possibly through the fecal transmission route. However, these bat influenza viruses poorly adapt to non-bat species like ferrets and mice (Ciminski et.al 2019). Certain mutants of the virus do replicate efficiently in mice, suggesting that evolutionary forces might result in spillover causing mutants (Fig.3).
So, how are we ‘helping’?
As humans, it’s in our nature to keep expanding our territory. And for this purpose, we have, sadly, deforested and converted swathes of greenery into residential or commercial areas. Over the past decade, since 2010, it has been estimated by the UN FAO that each year, about 10 million hectares of forest were cleared (https://ourworldindata.org/deforestation).The disturbances thus caused to nature result in the displacement of animal populations that have their niches in the disturbed areas. These also result from humans encroaching onto the niches of organisms like bats, that bring them together in the same ecological space in the first place.
It also may be due to farming or husbandry practices carried throughout generations, which makes certain population groups perpetually susceptible to certain diseases. While nature will keep throwing these curveballs at us in the form of ever-evolving viruses, we need to be mindful of our part in facilitating their spillover or spread, and take sufficient steps to curb them. While arthropod borne viruses are a major example of human spillover caused due to anthropogenic disturbances, influenza viruses, despite being zoonotic in origin, are not transmitted by arthropods like mosquitoes or ticks.
The more introductions of a virus into a new animal population, more will be the mutations and evolutionary changes in them, and so the migration of bat populations to more urban settings may enhance such a spread and the eventual adaptation of the virus to humans. It is not uncommon for fruit bat populations to make their homes in urban or peri-urban areas, thus acting as possible reservoirs of viruses hanging around dense human populations.
Age structure and migratory status of birds have been shown to influence the ecology of avian influenza viruses (Van Dijk et al.,2014). In a similar vein, the fragmentation of bat habitats can result in their populations, or roosts, dividing into smaller parts and settling in faraway locations, which may include urban and peri-urban areas. While it is lucrative to view a disease through a ‘one host-one pathogen’ lens, the reality may be far broader. With so many hosts available, not factoring in all of their roles may heavily undermine the potential these viruses possess. The understanding of viral diseases, therefore, warrants a more holistic approach towards these transient entities, for which concepts such as One Health have emerged (https://www.cdc.gov/onehealth/basics/index.html). One Health factors in the interconnection among people, animals, plants and the environment, thus making infectious disease research a collaborative effort among various sectors and disciplines.
A recent study by Bergener and colleagues in 2019 does focus on how local factors like livestock availability, climate and elevation along with population-specific attributes like age and genetic distance amongst the host impact the viral community. So, the more bat habitats are changed, more will their dietary habits be altered, switching to foods that may cause physiological stress, and thus more virus shedding. Fruit trees and paddy fields provide nutrition to bats that roost around agricultural land, so the shedding of urine and faeces in these areas can expose humans and farm animals to viral pathogens utilizing them as ‘vehicles’ of transmission.
Our arms against Influenza!
Faced with these curveballs nature throws at us – these seemingly genius viruses that are constantly shapeshifting to become ever mightier – what are our odds? A global initiative called FluNet aims to survey the timescape and occurrence of influenza cases in each year, starting from 1997, when it was first launched (https://www.who.int/tools/flunet). It collects data from reference centers under the Global Influenza Surveillance and Response System (GISRS). The GISRS has a global network of laboratories that collate and regularly report all the detected influenza cases.

The laboratories are arranged into hierarchies, starting from National Influenza Centres to WHO Collaborating Centres for Reference & Research on Influenza. Accumulating it into charts easily accessible by anyone on the internet. These show which species and subtypes of influenza are prevalent to what extent in each year, and also forms the basis on which predictions are made to formulate vaccines for each successive winter season, when influenza cases peak. These vaccines are recommended by the WHO every six months, based on which large scale manufacturers produce vaccine doses in case an epidemic or pandemic breaks out (Fig.4).
One can also observe from the FluNet charts that in 2020, while the entire world was quarantined due to COVID-19, influenza cases had dropped to all time lows, clearly indicating the benefit mask-wearing and quarantining brought about. Rigorous research is constantly ongoing into the nature of influenza virus mutations, including the slow ‘drift’ mutations and the massive ‘shift’ ones. These are but a few examples of the painstaking efforts investigators and healthcare workers are devoting towards understanding and keeping tabs on influenza. With the already available information, and the ongoing research, it suffices to say that while the battle may be far from over, we are always going to be armed and ready to fight.
Reference:
- Tong, Suxiang et al. “A distinct lineage of influenza A virus from bats.” Proceedings of the National Academy of Sciences of the United States of America vol. 109,11 (2012): 4269-74. doi:10.1073/pnas.1116200109
- Tong, Suxiang et al. “New world bats harbor diverse influenza A viruses.” PLoS pathogens vol. 9,10 (2013): e1003657. doi:10.1371/journal.ppat.1003657
- Karakus, Umut, et al. “MHC class II proteins mediate cross-species entry of bat influenza viruses.” Nature 567.7746 (2019): 109-112.
- Ciminski, Kevin, et al. “Bat influenza viruses transmit among bats but are poorly adapted to non-bat species.” Nature microbiology 4.12 (2019): 2298-2309.
- Bergner, Laura M., et al. “Demographic and environmental drivers of metagenomic viral diversity in vampire bats.” Molecular ecology 29.1 (2020): 26-39.
- Yang, Wenyu, Tony Schountz, and Wenjun Ma. “Bat Influenza Viruses: Current Status and Perspective.” Viruses 13.4 (2021): 547.
- van Dijk, Jacintha G B et al. “Juveniles and migrants as drivers for seasonal epizootics of avian influenza virus.” The Journal of animal ecology vol. 83,1 (2014): 266-75. doi:10.1111/1365-2656.12131

